
Introduction
Next-generation sequencing (NGS) operates as a precision workflow where fragmentation, adapter ligation, and sequencing chemistry each depend on the quality of what goes in. Yet labs routinely optimize library preparation protocols while treating extraction as a procedural checkbox—a disconnect that makes upstream nucleic acid quality the most overlooked source of NGS failure.
When sequencing runs produce low yields, uneven coverage, or variant call errors, most teams troubleshoot library prep kits or instrument calibration. The root cause is usually upstream: insufficient DNA, contaminated RNA, or degraded input that never met the minimum quality threshold for reliable library construction.
Extraction doesn't just precede library prep. It determines whether the downstream workflow can succeed at all.
That upstream dependency is what this article examines: which purity metrics affect adapter ligation efficiency, how integrity scores predict coverage uniformity, and why extraction QC gates prevent costly repeat runs before they happen.
TLDR
- Extraction quality controls every downstream NGS outcome: library complexity, variant call accuracy, and cost per usable result
- Three extraction parameters matter most: purity (A260/280 ratio), integrity (DIN/RIN score), and accurate fluorometric quantification
- Contaminated or degraded input reduces adapter ligation efficiency, inflates PCR bias, and generates unreliable variant calls
- Magnetic bead-based automation removes operator variability, ensuring consistent input quality across every run
- Upfront extraction QC prevents library prep failures and eliminates repeat sequencing runs
What Is NGS Library Preparation — And Why Starting Material Is Everything
NGS library preparation converts extracted nucleic acids into adapter-flanked fragments of known size that a sequencer can read. The library contains everything the sequencer will analyze—every fragment, every adapter, every molecular barcode. The sequencer reads only what the library contains. It cannot correct for upstream errors.
Extraction is Step 1 of every NGS workflow, sitting upstream of:
- Fragmentation
- Adapter ligation
- Size selection
- Library QC
Any quality compromise introduced at extraction—contamination, degradation, inaccurate quantification—is locked into all subsequent steps. If the input DNA contains phenol residues that inhibit ligase, adapter ligation fails regardless of how carefully you optimize the library prep protocol.
Extraction quality is a gating variable that determines whether you'll achieve analyzable, accurate reads per sample. Understanding what makes extraction succeed or fail—purity thresholds, yield consistency, integrity scores—is the starting point for any NGS workflow that needs to hold up downstream.
Key Advantages of High-Quality DNA/RNA Extraction for NGS Library Prep
The three advantages below address the most operationally significant impacts of extraction quality: library construction efficiency, sequencing data reliability, and overall workflow cost and throughput—each directly measurable in a lab setting.
Advantage 1: Higher Library Yield and More Efficient Adapter Ligation
High-purity DNA/RNA ensures that enzymatic steps in library prep—end repair, A-tailing, and adapter ligation—work at full efficiency. Purity is measured by A260/280 ratio, with accepted thresholds of ~1.8 for DNA and ~2.0 for RNA. Contaminants such as proteins, phenol residues, or salts directly inhibit these enzymes and reduce the proportion of fragments successfully ligated with adapters.
How this plays out in practice:
- EDTA concentrations > 1 mM cause tagmentation reactions to fail outright
- High salt or EDTA levels prevent T4 DNA Ligase from functioning
- Samples with contaminated or degraded input consistently produce lower-complexity libraries, requiring more PCR cycles to reach sequencing-ready concentrations—which amplifies duplicates and coverage bias
Clinical and operational impact:
More usable library molecules per nanogram of input is the direct result of clean extraction. This is especially critical for low-input samples—cfDNA, FFPE, biopsies—where there is no room to compensate for yield loss. Reducing input DNA from 50 ng to 10 ng compromises library complexity, negatively affecting variant detection sensitivity.
Higher yield from clean input means fewer PCR cycles needed, lower reagent consumption, and less likelihood of needing to repeat the library prep run. For reference, US genomics lab pricing puts library prep at $98–$142 per sample—every failed library is unrecoverable reagent waste regardless of geography.
KPIs impacted:
- Library complexity (unique molecule count)
- On-target read percentage
- Adapter ligation rate
- PCR cycle number required
- Reagent cost per successful library

When this advantage matters most:
This advantage is highest-impact when working with limited or precious samples—oncology biopsies, prenatal cfDNA, FFPE archival tissue, single-cell inputs—where input volume cannot simply be increased to compensate for inefficiency.
Advantage 2: More Accurate and Reproducible Sequencing Data
Nucleic acid integrity—the degree to which DNA is unfragmented (measured by DIN score) or RNA is undegraded (measured by RIN score)—directly controls the size distribution of library fragments and the uniformity of genome coverage. Highly degraded input skews fragment sizes toward short molecules, creating uneven depth across the target region and gaps at the sequencing stage.
How this plays out in practice:
RNA with a low RIN (below 7) introduced into an RNA-seq library prep produces a 3'-biased library where reads cluster at transcript ends rather than being evenly distributed. This makes gene expression quantification unreliable and differential expression analysis misleading.
For DNA, a DIN threshold of ≥ 3 is recommended for FFPE samples to ensure an on-target rate > 70% and coverage at 10× > 90%. Lower DIN correlates with decreased unique on-target reads and reduced average depth of coverage.
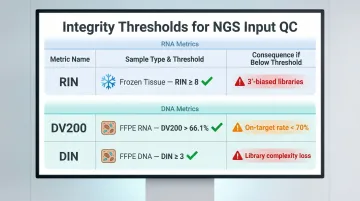
The key quality thresholds to track across sample types:
| Metric | Sample Type | Recommended Threshold | Impact if Below Threshold |
|---|---|---|---|
| RIN | Fresh/frozen tissue | ≥ 8 | 3'-biased libraries, unreliable expression data |
| RIN | General RNA-seq | ≥ 7 | Uneven transcript coverage |
| DV200 | FFPE RNA | > 66.1% | Insufficient NGS library yield |
| DV200 | FFPE RNA (minimum) | ≥ 30% | Not recommended for library prep |
| DIN | FFPE DNA | ≥ 3 | On-target rate drops below 70% |
Reproducibility across batches:
Consistent integrity scores across samples and batches eliminate a major source of inter-sample variability in NGS results. This is especially critical in clinical and diagnostic settings where sequencing data informs treatment decisions—variant calls, fusion detection, or copy number changes must be reproducible run-to-run.
Illumina recommends RIN ≥ 8 for optimal mRNA library prep. For FFPE RNA, DV200 > 66.1% strongly correlates with sufficient NGS library yield. Samples with DV200 < 30% should not proceed to library prep.
KPIs impacted:
- Uniformity of coverage (% bases above threshold depth)
- Variant call accuracy and sensitivity
- False-positive/false-negative rate in variant detection
- Inter-batch reproducibility
- RIN/DIN score as a quality gate metric
When this advantage matters most:
This is most critical in applications requiring high sensitivity—somatic variant detection at low allele frequency (liquid biopsy, ctDNA), RNA expression profiling, and fusion gene detection—where data quality directly affects clinical or research conclusions.
Advantage 3: Lower Cost Per Usable Result and Fewer Failed Runs
Sequencing reagents, library prep kits, and sequencing run time represent substantial per-sample costs. When extraction quality is insufficient and libraries fail QC or produce unusable data, these costs are not recovered—the sample must be reprocessed or the run is wasted.
How this plays out operationally:
Labs that do not standardize extraction QC often discover library failures only after committing to sequencer time. Failed runs create backlog, delay turnaround times, and in clinical settings can compromise patient care timelines. US reference pricing gives a sense of the exposure: a NovaSeq SP run costs $5,175–$6,538 and a NovaSeq X Plus 10B lane runs $1,823—costs that scale proportionally in any high-throughput setting.
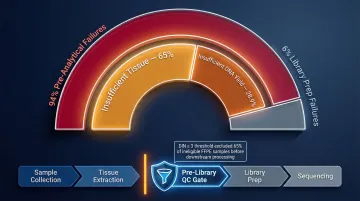
In a clinical oncology study of 1,528 samples, 22.45% failed NGS testing—94% due to pre-analytical factors: insufficient tissue (65%) or insufficient DNA yield <100 ng (28.9%). Only 6.1% of failures occurred during the analytical library preparation phase itself.
How pre-library QC gates change the economics:
Labs implementing pre-library QC gates—minimum concentration thresholds, purity checks, and integrity scoring—catch failures before expensive downstream steps are initiated. Applying a DIN ≥ 3 threshold for FFPE samples allowed one facility to exclude 65% of samples (488 out of 751) from downstream processing, saving both sequencing time and reagent costs on those samples.
As throughput scales, the compounding cost of poor extraction quality grows proportionally. Standardising extraction quality at the front end protects the return on investment in sequencing infrastructure.
KPIs impacted:
- Cost per usable sequencing result
- Percentage of library prep runs passing QC on first attempt
- Average turnaround time from sample receipt to reportable result
- Repeat run rate
When this advantage matters most:
This advantage is highest at high-throughput facilities—clinical diagnostic labs, cancer genomics centers, biobanks, NIPT labs—where run failure rates multiply across hundreds of samples and reagent waste becomes a significant budget line.
What Happens When Extraction Quality Is Ignored
When labs skip or deprioritize extraction QC, inhibitor carryover silently reduces enzyme activity in library prep, leading to low-yield or adapter-dimer-dominated libraries that fail pre-sequencing QC. The failure mode is often misattributed to the kit or the sequencer rather than the sample itself.
Additional real-world consequences:
- RNA degradation in samples not processed quickly or stored incorrectly leads to biased transcriptomic data
- gDNA contamination in RNA preps introduces false signals in gene expression studies
- Inconsistent extraction protocols between operators or batches create inter-run variability that undermines study reproducibility
- Severely fragmented FFPE DNA with deamination generates false-positive mutation calls—specifically C:G > T:A transitions
The compounding costs add up quickly:
- Higher reagent waste from failed library prep runs
- Longer turnaround times when samples must be reprocessed
- Misdiagnosis risk in clinical settings where result accuracy is non-negotiable
- Scaling difficulties when extraction variability compounds across batches
Each of these failures traces back to a fixable upstream problem — and QC checks at the extraction stage catch them before they reach the sequencer.
How to Ensure Extraction Quality Meets NGS Standards
Labs should verify minimum QC parameters before proceeding to library prep:
Required QC checks:
| QC Method | Purpose | Acceptance Criteria |
|---|---|---|
| UV Spectrophotometry | Purity assessment | A260/280: ~1.8 (DNA), ~2.0 (RNA) |
| UV Spectrophotometry | Contaminant detection | A260/230: 2.0–2.2 |
| Fluorometry (Qubit) | Accurate quantification | Kit-specific input mass |
| Microfluidics (TapeStation) | Integrity assessment | DIN ≥ 3 (FFPE DNA), RIN ≥ 8 (RNA) |
Fluorometric vs. UV Quantification
Illumina and CLSI guidelines explicitly recommend fluorometric quantitation (e.g., Qubit) over UV spectrophotometry (e.g., NanoDrop) for NGS. UV methods measure total nucleic acids and impurities, overestimating amplifiable DNA/RNA. Fluorometric methods use target-specific dyes that bind only dsDNA, providing accurate measurements even at low concentrations.
A260/230 ratios: A ratio of 2.0–2.2 is generally acceptable. Lower values indicate contamination by phenol, guanidine HCl, guanidine thiocyanate, or carbohydrates, which inhibit enzymatic reactions.
Standardization and Automation
Manual, variable extraction introduces batch effects that are invisible in single-sample QC but emerge as reproducibility problems at scale. Instrument, reagent lot, and operator procedure all need to be locked down consistently across runs.
Automated magnetic bead-based extraction systems address this directly. They produce consistent purity and yield, eliminate operator-dependent variability, and generate an auditable extraction record. Automated systems retain the accuracy of manual extraction while enabling higher throughput without compromising DNA integrity or downstream NGS read quality.

Cambrian Bioworks' Manta, a CE-IVD certified automated DNA extraction system, is designed for clinical NGS workflows. It completes extraction in approximately 30 minutes with minimal genomic DNA contamination, processing 1–32 samples per run with no batching constraints.
Conclusion
Extraction quality controls the outcome of every NGS run downstream. What the sequencer receives, it cannot fix — degraded input, inhibitor carryover, and inaccurate quantification all propagate forward into failed libraries, distorted variant calls, and wasted reagent costs.
The advantages of high-quality extraction—better library yield, more accurate data, and lower cost per result—compound over time and at scale. In clinical oncology, 94% of NGS failures trace back to pre-analytical extraction problems, not library prep or instrument errors. Implementing extraction QC gates catches these failures before expensive downstream steps begin.
Purity, integrity, and quantification accuracy are set at extraction — not recoverable afterward. Building these as defined, repeatable checkpoints into every NGS workflow is what separates labs that consistently produce reportable results from those that troubleshoot at the sequencing stage. Automating extraction standardizes all three parameters across operators, sample types, and run volumes, making quality a system property rather than an individual judgment call.
Frequently Asked Questions
What is library preparation in next generation sequencing?
NGS library preparation is the process of converting extracted DNA or RNA into adapter-flanked, size-selected fragments ready for sequencing. Library quality depends directly on the purity, integrity, and concentration of the input nucleic acid—poor extraction compromises every downstream step.
What are the steps in next generation sequencing?
The four main NGS workflow steps are: (1) nucleic acid extraction, (2) library preparation (fragmentation, adapter ligation, size selection, QC), (3) sequencing, and (4) data analysis. Step 1 sets the quality ceiling for all that follows—extraction errors cannot be corrected downstream.
How long does it take to prep a library for NGS?
Library prep typically takes 1.5 to 6+ hours depending on the kit and application, but this timeline assumes extraction has already been completed and QC-passed. Poor extraction quality can add hours or days if repeat extraction is needed.
How long does sequencing take to process?
Sequencing run times range from a few hours to several days depending on the platform and depth required. In practice, total time-to-result is most often delayed by upstream library quality failures, not the sequencer itself.
What is Illumina's next generation sequencing workflow?
Illumina's NGS workflow uses sequencing by synthesis across the same four-step structure: extraction, library prep, cluster generation, and data analysis. Illumina recommends UV spectrophotometry and fluorometric QC at the extraction stage before any library prep begins.
What quality metrics should DNA or RNA meet before NGS library preparation?
Key pre-library QC metrics include:
- A260/280 ratio — purity assessment
- A260/230 ratio — solvent contamination check
- Fluorometric concentration (Qubit preferred over UV absorbance)
- Integrity score — DIN for DNA, RIN for RNA
Minimum thresholds vary by application (DIN ≥ 3 for FFPE DNA; RIN ≥ 8 for mRNA). Any sample failing these checks should not proceed to library prep.


