
Introduction
Pathology archives hold millions of FFPE tissue blocks — decades of clinically annotated samples representing some of the most complete oncology records in existence. Yet the same fixation process that preserves tissue morphology makes RNA notoriously difficult to work with: fragmented, chemically modified, and resistant to standard sequencing workflows.
For researchers pursuing gene expression profiling or fusion gene detection from archival tissue, this creates a real tension.
The core challenge is two-fold: formalin cross-linking degrades RNA integrity, and rRNA content can overwhelm sequencing reads if depletion is inadequate. A suboptimal extraction or depletion step doesn't just lower data quality — it can render an irreplaceable archival sample unusable. Getting the workflow right the first time matters.
This guide covers best practices for each stage of the FFPE RNA-seq workflow—from tissue sectioning and extraction to RNA quality assessment, rRNA depletion strategy selection, and library preparation considerations.
TLDR
- FFPE RNA is inherently degraded and chemically modified; standard RNA-seq protocols for fresh or frozen samples often fail
- DV200 >30% is the minimum threshold for RNA-seq; RIN is not informative for FFPE samples
- Use hybridization-based RNase H depletion for rRNA removal; it outperforms bead-capture on degraded FFPE RNA
- Poly-A enrichment should be avoided for FFPE RNA due to severe 3' bias and transcript loss
- Fluorometric quantification (Qubit) with DNase treatment is required to prevent inaccurate RNA input
Why FFPE RNA Is Uniquely Challenging for Transcriptomic Studies
The Chemistry of Formalin Fixation
Formaldehyde generates covalent cross-links between nucleic acids and proteins, creating DNA-RNA and DNA-protein bonds that must be reversed during extraction. This alone complicates every downstream step.
Beyond cross-linking, formalin introduces cytosine deamination and RNA fragmentation during the prolonged heat steps of tissue processing. These chemical changes set FFPE RNA apart from fresh-frozen material — and set realistic expectations for what any extraction protocol can recover.
Cumulative Degradation Factors
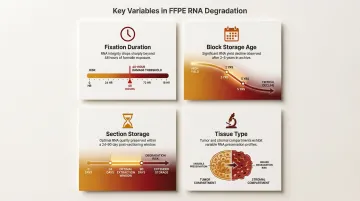
Multiple variables compound RNA damage in FFPE tissue:
- Fixation duration: Over-fixation beyond 48 hours dramatically increases damage
- Block storage age: RNA amplification success declines significantly after 2–5 years of storage
- Section storage after cutting: Process sections within 24–90 days to minimize degradation
- Tissue type: Tumor regions exhibit different RNA integrity profiles compared to stromal areas
Downstream Consequences of Poor Quality
Compromised FFPE RNA produces predictable sequencing failures. Each one erodes data quality in a different way:
- High intronic read rates from incomplete transcript coverage
- Elevated residual rRNA consuming usable sequencing depth
- Low mapping efficiency due to fragmented, modified reads
- Duplication artifacts inflating apparent expression levels
- Biased gene expression quantification skewing biological conclusions
Getting extraction and ribosomal depletion right is the only way to prevent these failures before they reach your sequencer.
Best Practices for FFPE RNA Extraction
Every pre-analytical variable—from sectioning to elution volume—determines whether library preparation succeeds or fails.
Sectioning and Tissue Input
- Use 5–10 μm thick sections rather than a single thick section
- Discard the first 3–4 sections to remove surface degradation
- Process sections immediately or store sealed at –20°C for no more than 90 days
- Use 1–8 sections depending on tissue surface area and cellularity
Pathologist-guided macrodissection or microdissection enriches for tumor-rich regions before extraction. Avoid necrotic areas, melanin-rich zones, and non-target immune infiltrates that reduce signal quality and inflate variability.
Deparaffinization and Digestion
Deparaffinization:
- Xylene wash followed by ethanol washes and complete drying is the standard baseline
- Acceptable alternatives include heptane and proprietary buffer-based methods
- Avoid microwave heating and solvents like HemoD for RNA applications
- Incomplete deparaffinization is a common failure point
Proteinase K digestion and demodification:
- Digest at 55–56°C for 15 minutes to overnight (kit-dependent)
- Demodify at 80°C for 15 minutes to reverse formalin-induced RNA modifications
- Over-demodification (>15 min) reduces DV200 and should be avoided
Extraction Method and Automation
- Silica membrane-based kits and magnetic bead-based kits are most reliable
- Avoid resin-filter and heat-only extraction methods
- Organic extraction (phenol-chloroform) can be acceptable but is less consistent
When running multiple FFPE samples, automated magnetic bead-based platforms significantly reduce batch-to-batch variability. They also enforce consistent elution volumes—a detail that directly affects downstream quantification accuracy and library yield.
Assessing FFPE RNA Quality: Key Metrics and Thresholds
Why RIN Fails for FFPE RNA
RIN (RNA Integrity Number) is not a useful quality metric for FFPE RNA. Ribosomal peaks are not discernible in degraded samples, making RIN scores either unmeasurable or misleading. Never use RIN as a pass/fail criterion for FFPE RNA-seq workflows.
Instead, DV200 is the accepted standard for evaluating FFPE RNA fitness for sequencing.
DV200: The Appropriate Quality Metric
DV200 measures the percentage of RNA fragments greater than 200 nucleotides in length, assessed by microfluidic capillary electrophoresis (Bioanalyzer or Fragment Analyzer on an RNA Pico chip).
Standard thresholds:
- DV200 >30%: Minimum accepted threshold for most RNA-seq platforms
- DV200 30–50%: Increase input amounts to compensate for lower fragment integrity
- DV200 <30%: Sample is too degraded for reliable RNA-seq; consider alternative library prep strategies or exclude from sequencing
Quantification Best Practices
Use fluorometric methods (Qubit RNA HS assay) rather than spectrophotometry. NanoDrop measurements overestimate FFPE RNA yield by approximately 32% due to co-absorbing contaminants like fragmented genomic DNA, making spectrophotometry unreliable for standardizing input amounts.
Critical steps:
- Include DNase treatment before quantification to avoid DNA contamination inflating yield measurements
- DNase treatment eliminates the NanoDrop–Qubit discrepancy, achieving correlation coefficients of R=0.91–0.99, according to a 2023 Lab Investigation study
- For low-input workflows (5–10 ng), accurate quantification is critical to hit kit-specific input ranges
Ribosomal RNA Depletion: Strategies and Comparisons
Ribosomal RNA typically constitutes over 80% of total cellular RNA. Without depletion, the vast majority of sequencing reads map to rRNA rather than informative transcripts, wasting sequencing depth.
Poly-A enrichment cannot substitute for rRNA depletion in FFPE RNA — degraded RNA lacks intact poly-A tails, resulting in severe 3' bias and poor transcript coverage.
Three main depletion strategies are available, each with distinct trade-offs in sensitivity, residual rRNA, and suitability for degraded FFPE inputs.
Hybridization-Based Depletion (Pre-cDNA, Bead-Capture)
Single-stranded DNA probes hybridize to cytoplasmic (5S, 18S, 28S, 5.8S) and mitochondrial (12S, 16S) rRNA, which are then captured on magnetic beads and removed.
Performance characteristics:
- Effective for intact, high-quality RNA
- Residual rRNA >10% on degraded FFPE RNA
- Kits like Illumina Stranded Total RNA Prep with Ribo-Zero Plus achieve ~0.1% residual rRNA in high-quality RNA but perform variably on degraded samples
Hybridization-Based Depletion (Pre-cDNA, RNase H-Mediated)
DNA probes hybridize to rRNA sequences and RNase H degrades the RNA strand of the resulting RNA:DNA hybrid, followed by DNase I to remove DNA probes.
Performance characteristics:
- Effective across a wide range of RNA quality levels, including 1-year and 10-year-old FFPE RNA
- Does not rely on intact RNA structure
- Lower residual rRNA percentage than bead-capture on degraded samples
Post-cDNA rRNA Depletion
Used in kits like TaKaRa SMARTer Stranded Total RNA-Seq Kit v2, rRNA removal occurs at the cDNA level using specific elimination probes after reverse transcription.
Performance characteristics:
- Retains higher transcript diversity from degraded inputs
- Well-suited for low-input FFPE samples (as low as 10 pg)
- Higher residual rRNA reads (~17%) requiring greater sequencing depth
- Fragmented RNA is converted to cDNA before depletion
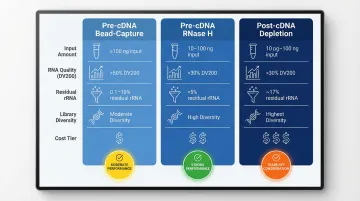
Comparison Table
The table below summarizes how each approach compares across key decision criteria.
| Approach | Input RNA Range | DV200 Threshold | Residual rRNA | Transcript Diversity | Cost |
|---|---|---|---|---|---|
| Pre-cDNA Bead-Capture | ≥100 ng | >50% | 0.1–10% | Moderate | ₹₹ |
| Pre-cDNA RNase H | 10–100 ng | >30% | <5% | High | ₹₹ |
| Post-cDNA Depletion | 10 pg–100 ng | >30% | ~17% | Highest | ₹₹₹ |
Putting It All Together: An FFPE RNA-Seq Best Practices Workflow
End-to-End Sequential Framework
Follow this decision framework for reliable results:
- Evaluate block quality and select appropriate tissue sections
- Deparaffinize and perform proteinase K digestion with demodification
- Extract RNA using validated silica or magnetic bead-based method
- Quantify by fluorometry and assess DV200 by Bioanalyzer
- Choose rRNA depletion strategy based on DV200 and input availability
- Proceed to library preparation
Each step requires kit-specific experimental validation before applying to a clinical cohort.
rRNA Depletion Method Selection Guide
For DV200 >50% and ≥100 ng input:
- Either pre-cDNA bead-capture or RNase H methods are appropriate
For DV200 30–50% or limited input (10–100 ng):
- RNase H-based or post-cDNA depletion is preferred
For DV200 <30%:
- Higher input amounts should be used if possible
- Post-cDNA approaches or specialized low-input kits are recommended
Once depletion strategy is confirmed, library preparation choices determine how well the resulting data holds up to downstream analysis.
Library Preparation and Sequencing Considerations
- Stranded library prep: Preserves transcriptional directionality — critical for accurate isoform and antisense transcript detection
- Sequencing depth: Plan for higher read requirements than fresh-frozen RNA, particularly when using post-cDNA depletion, where rRNA carry-through can inflate total read counts
- Unique molecular identifiers (UMIs): Reduce PCR duplication artifacts, which are elevated in degraded-input libraries
- Automation: Automated liquid handling reduces reagent variability and supports consistent throughput across larger sample batches
Key Principle
Mismatched combinations are the most common cause of library failure. Poly-A enrichment on DV200 45% FFPE RNA, or bead-capture depletion on a 10-year-old block, will produce low-complexity libraries regardless of downstream sequencing depth.
A structured, validated workflow protects archival FFPE samples — material that cannot be replaced — and delivers reproducible, clinically relevant transcriptomic data.
Frequently Asked Questions
What is FFPE RNA?
FFPE RNA refers to RNA extracted from formalin-fixed paraffin-embedded tissue blocks, the standard format for archival tissue storage in pathology. While these samples are abundant and clinically valuable, formalin fixation fragments and chemically modifies RNA, making it more challenging to use for transcriptomic studies than RNA from fresh or frozen tissue.
Does formalin destroy RNA?
Formalin does not destroy RNA outright, but causes significant chemical damage — including protein cross-links, cytosine deamination, and progressive fragmentation accelerated by high-temperature paraffin embedding and long storage. RNA from FFPE tissue is degraded but remains usable for RNA-seq with appropriate protocols.
How do you evaluate RNA quality?
FFPE RNA quality is assessed using DV200 (the percentage of fragments greater than 200 nucleotides), measured via capillary electrophoresis on a Bioanalyzer or Fragment Analyzer. RIN is not informative here because ribosomal peaks used to calculate it are not intact in FFPE samples. A DV200 above 30% is the minimum threshold for RNA-seq.
What is RNA depletion?
RNA depletion (specifically ribosomal RNA depletion) removes abundant rRNA from a total RNA sample before library preparation. Because rRNA comprises over 80% of total RNA, skipping this step means most sequencing reads map to rRNA rather than informative mRNA or non-coding transcripts — making depletion essential for whole-transcriptome RNA-seq.
What is the difference between poly A and rRNA depletion?
Poly-A selection enriches for polyadenylated mRNA by capturing transcripts via their poly-A tails using oligo-dT beads, while rRNA depletion uses probe hybridization to specifically remove ribosomal RNA sequences while retaining all other RNA species. For FFPE RNA-seq, rRNA depletion is strongly preferred because degraded RNA lacks intact poly-A tails, making poly-A selection inefficient and biased toward 3' transcript ends.


